An important consideration in the field of leukemia stem cells (LSCs) is to define culture condition that best mimic the in vivo microenvironment. LSCs rapidly differentiate when introduced in culture. This hampers our ability to conduct cell-based high-throughput lethality screens and partly explains the lack of success in drug development for this disease.
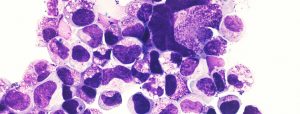
To overcome these constraints we conducted a high-throughput screening and identified small molecules that inhibit differentiation and support LSC activity in vitro. The first class of compounds identified, represented by SR1, inhibits the aryl hydrocarbon receptor (AhR) pathway. Interestingly this pathway is rapidly upregulated when cells are introduced in culture with obvious and rapid cell differentiation. A second compound, UM729, in combination with SR1 was shown to better maintain LSC activity in vitro. UM729 lacks AhR suppressor activity, which indicates that at least 2 different pathways are involved. These 2 compounds define the best culture conditions for improved ex vivo culture of primary human AML cells (Pabst et al., Nature Methods 2014).
Unfortunately, approximately 30% of specimens respond poorly to these molecules suggesting that a 3rd pathway is operational in securing LSC self-renewal divisions. That is why we continue to screen new molecules and investigate our best hits.
 The phenotypical differences arising from minor variations at the molecular level could have major effect on responses of leukemias to drugs. To address this issue we are screening the in vitro biological responses of primary human acute myeloid leukemia (AML) cells to various collections of chemical compounds in a media that transiently inhibits differentiation and supports leukemia stem cell activity ex vivo.
The phenotypical differences arising from minor variations at the molecular level could have major effect on responses of leukemias to drugs. To address this issue we are screening the in vitro biological responses of primary human acute myeloid leukemia (AML) cells to various collections of chemical compounds in a media that transiently inhibits differentiation and supports leukemia stem cell activity ex vivo. encountered with conventional therapies.
encountered with conventional therapies. Our role is to develop or assist in the development of predictive tools that will make use of clinical observations and chemogenomic measures of patient samples to provide guidance to clinicians in selecting treatment. For this, we favor a two-pronged approach.
Our role is to develop or assist in the development of predictive tools that will make use of clinical observations and chemogenomic measures of patient samples to provide guidance to clinicians in selecting treatment. For this, we favor a two-pronged approach. Current prognostic classification of Acute Myeloid Leukemia (AML) is based on cytogenetics and a limited number of mutations, where patients belong either to favorable, intermediate and adverse cytogenetic groups. The majority of patients will be classified into intermediate cytogenetics, an heterogenous group in which clinical outcome is highly variable and for which clinical decisions are difficult. This problem still leads to over- or under-therapy with dire consequences. In addition, a subset of patients with adverse cytogenetics will fail to respond to current therapies. More discriminatory risk stratification will lead to more accurate clinical decisions.
Current prognostic classification of Acute Myeloid Leukemia (AML) is based on cytogenetics and a limited number of mutations, where patients belong either to favorable, intermediate and adverse cytogenetic groups. The majority of patients will be classified into intermediate cytogenetics, an heterogenous group in which clinical outcome is highly variable and for which clinical decisions are difficult. This problem still leads to over- or under-therapy with dire consequences. In addition, a subset of patients with adverse cytogenetics will fail to respond to current therapies. More discriminatory risk stratification will lead to more accurate clinical decisions.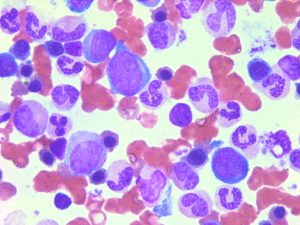 After induction or consolidation chemotherapy, patients in morphological complete remission can still have residual leukemic cells (i.e. minimal residual disease, MRD). MRD positivity is associated to an increased risk of relapse and a poorer clinical outcome. Currently, MRD is tested either by flow cytometry (leukemia- associated immunophenotype) or by RT-qPCR (aberrantly expressed gene, fusion transcript or mutation). However, a good MRD test is lacking in several AML subgroups.
After induction or consolidation chemotherapy, patients in morphological complete remission can still have residual leukemic cells (i.e. minimal residual disease, MRD). MRD positivity is associated to an increased risk of relapse and a poorer clinical outcome. Currently, MRD is tested either by flow cytometry (leukemia- associated immunophenotype) or by RT-qPCR (aberrantly expressed gene, fusion transcript or mutation). However, a good MRD test is lacking in several AML subgroups.
Research resources and scientific data generated through the Leucegene project are made accessible to the scientific community after publication through international repositories.
The Leucegene RNA sequencing dataset (691 AML samples) is available in the Gene Expression Omnibus repository (GEO accession number GSE232130). Non-identifiable clinical data for the Leucegene cohort are available to academic investigators for a research project in hematological cancers approved by a research ethics committee in accordance with the procedures of the Quebec Leukemia Cell Bank: Banque de cellules leucémiques du Québec – Request for Cells/Data (bclq.org).
 RNA sequencing data is accessible through GEO or SRA.
RNA sequencing data is accessible through GEO or SRA.
More specifically, the following datasets are available:
- Leucegene: AML sequencing – Accession # GSE49642.
- Leucegene: AML sequencing (part 2) – Accession # GSE52656.
- Leucegene: AML sequencing (part 3) – Accession # GSE62190.
- Leucegene: AML sequencing (part 4) – Accession # GSE66917.
- Leucegene: AML sequencing (part 5) – Accession # GSE67039.
- Leucegene: AML sequencing (part 6) – Accession # GSE106272.
- Leucegene: AML sequencing– Accession # GSE67040 (SuperSerie containing the 6 SubSeries above).
- Leucegene: ALL sequencing – Accession # GSE49601.
- Transcriptome of Primitive Human Hematopoietic Cells: A New Resource to Find hHSC-Specific Genes – Accession: # GSE48846 and #GSE51984.
- Transcriptome analysis of G protein-coupled receptors in distinct genetic subgroups of AML – Accession: GSE98310 .
- Whole exome sequencing of human leukemia: SRA – Accession: PRJNA358716.

 AML patient samples have been obained from the
AML patient samples have been obained from the